
Inspection readiness in clinical research is the condition that exists when every site within a research network can produce complete, accurate, and current evidence of research activity at any point in the study lifecycle — without requiring emergency preparation, manual record reconstruction, or document requests sent to individual teams.
This guide explains what inspection readiness means in practice, why the standard definition fails in a multi-site CRDC environment, what MHRA inspectors actually examine when they visit a hub or spoke site, and how AQ supports CRDC networks with the connected documentation, quality governance, and operational control that makes inspection readiness a continuous operating condition rather than a periodic event.
What you will understand through AQ’s Guide to Inspection Readiness in Clinical Research:
- Inspection readiness is a continuous operating condition, not a last-minute preparation task.
- CRDC hubs are accountable for inspection readiness across every participating site.
- MHRA inspects compliance, authorised activity, and contemporaneous evidence at any site, at any time.
- Understand the most common MHRA inspection findings across NHS research organisations.
- Learn why pre-inspection preparation falls short of ALCOA+ expectations.
- See why documentation, delegation, quality, and pharmacy require continuous oversight.
- Discover how fragmented systems create hidden inspection risks across CRDC networks.
- Understand how real-time cross-site visibility strengthens governance and inspection readiness.
- See how eISF, ePSF, CAPA, Digital DoA, and QMS work together as one connected governance platform.
- Learn how AQ helps CRDCs maintain continuous inspection readiness with a live cross-site dashboard.
What is Inspection Readiness in Clinical Research?
Inspection readiness is the state a clinical research site or network reaches when it can demonstrate, at any moment and without advance reconstruction, that research has been conducted in compliance with the applicable regulatory framework, the approved protocol, and good clinical practice.
It is not a checklist that gets completed before an inspector arrives. It is not a filing sprint triggered by a monitoring visit notification. It is not a document review programme that runs once a quarter. Inspection readiness is a continuous operational condition — the product of research governance being under active, structured control throughout the life of every study.
The Medicines and Healthcare products Regulatory Agency (MHRA) conducts GCP inspections of clinical trial sponsors, investigator sites, contract research organisations, and other parties involved in the conduct of clinical trials of investigational medicinal products. Notably, an inspection can be routine and risk-based, triggered by a reported serious breach, or initiated based on intelligence from other regulators or internal MHRA findings. Organisations have no guarantee of advance notice sufficient to prepare from a standing start.
The inspector doesn’t inspect a filing system. An inspector examines evidence — to ensure that research was conducted correctly, that decisions were made by authorised people, that participants were protected, that data reflects what actually happened, and that any deviation from the expected process was formally identified, investigated, and resolved before the inspector arrived.
Inspection readiness is the condition that makes that evidence visible, current, and complete at any moment — not the week before a visit.
Also Read: Clinical Research Audits: Types, Process, Checklist, and Audit Readiness Guide
Why Inspection Readiness Means Something Different in a CRDC Network?
A single research site has one investigator site file, one delegation log, one pharmacy record, and one quality history. A CRDC network running a study across eight participating sites has eight of each. The hub is accountable for governance across all of them.
CRDCs operate under MHRA regulatory oversight. The hub — the lead NHS trust — holds governance accountability for network-wide research standards, document distribution, quality assurance, and sponsor engagement. Each participating spoke site retains its own research governance responsibilities while operating within the framework the hub maintains.
This structure creates an inspection risk that does not exist for single-site operations. MHRA can inspect the hub or any spoke site. What the inspector finds at a spoke site reflects on the hub’s governance, not just the site’s operations. A documentation gap at Spoke Site 4 is a hub accountability issue. An unresolved CAPA at Spoke Site 7 is a pattern risk the hub was responsible for detecting and acting on.
Inspection readiness in a CRDC network therefore requires something that single-site readiness does not: the hub must maintain current awareness of the documentation status, delegation currency, quality activity, and pharmacy accountability at every participating site — continuously, not periodically.
| Readiness Requirement | Single Site | CRDC Hub-and-Spoke Network |
|---|---|---|
| Investigator site file completeness | One site binder maintained locally | Eight or more per-site binders, each complete and current, visible to the hub in real time |
| Document version currency | One team managing one version set | Same current version confirmed at every participating site, with distribution and acknowledgement records |
| Delegation of authority records | One delegation log at one site | Current delegation records at every site, confirmed by the hub without requiring monitoring visits |
| CAPA status | Quality issues managed at site level | Deviations at any site visible to the hub, investigated with coordinated oversight, tracked to verified closure |
| Pharmacy accountability | One pharmacy record at one site | IP accountability records at each spoke, visible alongside ISF records within one hub governance view |
| Training and competency records | Site team manages its own training records | Training compliance confirmed across distributed teams, linked to the procedure version in effect at the time |
| Inspection evidence availability | Site team prepares from its own records | Hub can confirm readiness across every site without manual outreach or record reconstruction at any participating site |
What MHRA Inspectors Examine at a CRDC Site?
Understanding what inspectors look for is the starting point for understanding what inspection readiness actually requires. MHRA GCP inspections are not documentation reviews. They are evidence examinations. Inspectors examine whether the records confirm that research was conducted correctly — and whether any gap in the records signals a gap in the conduct of the trial itself.
Across NHS trust inspections, the most persistent categories of findings include the following.
Sponsor Oversight Failures
Sponsor oversight findings are among the most serious categories identified at NHS trust inspections. Nottingham University Hospitals, inspected by MHRA in March 2024 in its first GCP inspection in ten years, received two critical findings — one directly related to sponsor oversight of clinical trials, with inadequacies identified across quality assurance processes, monitoring, risk assessments, and essential documentation. The CAPA response required 62 corrective actions and 68 preventive actions.
In a CRDC context, sponsor oversight failures arise at the hub level when the hub cannot demonstrate real-time visibility of what is happening at participating sites. An inspector examining hub governance records expects to see evidence that the hub actively monitored site compliance, identified gaps, and responded to them. Evidence produced for the first time during inspection preparation does not satisfy that expectation.
Trial Master File and Investigator Site File Deficiencies
TMF and ISF deficiencies are consistently among the most frequent findings in MHRA GCP inspections. Recurring issues include incomplete files, poor version control, missing essential documents, inadequate audit trails, and retrospective data entry. MHRA emphasises ALCOA+ principles — Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available — as the standard against which documentation is assessed.
For a CRDC network, ISF deficiencies at spoke sites are not confined to those sites in inspection terms. The hub’s governance records must demonstrate that document completeness and version currency were actively monitored across every participating site. An inspector who finds an out-of-version consent form at a spoke site will ask when the hub governance team last confirmed the site’s document status. If the answer involves a monitoring visit that occurred several months ago, the governance gap is at the hub, not only the site.
Also Read: Electronic Investigator Site File (eISF) in Clinical Research
Delegation of Authority Gaps
Delegation records confirm that each research task was performed by a person with documented authority to perform it, assigned by the Principal Investigator, within the scope of their competence, at the time the task was carried out. Delegation gaps arise when role assignments are not updated after staff changes, when effective dates are unclear or missing, or when the delegation log does not reflect the tasks actually being performed by each team member.
In a CRDC network, delegation governance is a hub oversight responsibility. The hub cannot confirm that research at each spoke was conducted under current and correct authority without access to current delegation records at every site. An inspector examining a spoke site will cross-reference the delegation log against study activity records. A delegation gap that predates the last monitoring visit by several months signals that hub oversight of delegation governance was not continuous.
Also Read: What is DOA: Delegation of Authority in Clinical Research
Informed Consent Process Failures
Consent findings arise when participants were consented using a superseded version of the participant information sheet or consent form, when consent was obtained by a person without documented authority to take consent, when the timing of consent relative to study procedures cannot be confirmed, or when the consent record is incomplete.
Consent form version control is a specific vulnerability in multi-site studies. When a protocol amendment generates an updated participant information sheet, every participating site must receive, acknowledge, and file the current version before consenting any further participants. In a network where document distribution runs through email and shared drives, the hub has no structured mechanism to confirm that each site is operating on the current version at any given moment. Version drift accumulates between monitoring visits. An inspector examining consent records against protocol versions will detect it.
CAPA Process Deficiencies
CAPA findings at inspection arise when deviations were not formally documented, when investigations lack evidence of genuine root cause analysis, when corrective actions address the symptom rather than the cause, when preventive actions are absent, or when closure evidence is insufficient to confirm that the issue was actually resolved. Repeated findings of the same type — at the same site or across sites — signal that the CAPA process failed to identify and address the structural condition behind the events.
In a CRDC network, CAPA deficiencies compound when the hub quality team lacks visibility of quality events at spoke sites. A deviation at Spoke Site 2 and a related near-miss at Spoke Site 5 may represent the same root cause operating across the network. If each site manages its own CAPA in a local spreadsheet, the hub quality team cannot cross-reference events, detect the pattern, or extend preventive actions network-wide. An inspector who surfaces the pattern during inspection will find that the hub’s quality governance failed to identify it first.
Pharmacy and Investigational Product Accountability
Pharmacy findings at inspection arise when IP receipt, storage, dispensing, and return records cannot be reconciled, when storage condition documentation is incomplete or unverified, when dispensing events cannot be linked to specific participants and visits, or when pharmacy authorisations are expired or absent. Under UK Clinical Trials Regulations and MHRA GCP expectations, IP accountability is a regulatory requirement throughout the study lifecycle — not only at close-out.
Also Read: What is ePSF in Clinical Trial Data Management?
Why Pre-Inspection Preparation is a Governance Failure?
Most clinical research sites approach inspection readiness as a preparation task. When a monitoring visit is scheduled or an inspection notification arrives, teams review their site files, reconcile document placeholders, chase outstanding signatures, and collate evidence of CAPA resolution. The preparation takes days. The records produced may be complete at the moment of inspection. They were not complete at the moment the research activity occurred.
This distinction matters because ALCOA+ requires that records be contemporaneous: created at the time the activity occurred, reflecting what actually happened and when. A document signed the week before a monitoring visit to demonstrate that a procedure was followed several months ago is not a contemporaneous record. An inspector who examines the audit trail — when the document was uploaded, when it was signed, and how the timing relates to the activity it documents — will identify the gap.
Pre-inspection preparation that reconstructs scattered records does not produce inspection-ready evidence. It produces evidence that looks complete at the time of inspection but fails the contemporaneous test. Research teams that prepare intensively for every monitoring visit and inspection have identified the wrong problem. The problem is not that the preparation takes too long. The problem is that the research was not under continuous governance control throughout its delivery.
Inspection readiness built through continuous operational control looks different. Documents are filed at the time they are created. Signatures are obtained at the time the document is approved. Delegation records are updated when roles change. Deviations are raised when they occur. CAPAs are investigated before the next monitoring visit, not during it. The evidence exists because the work was done correctly — not because it was assembled for review.
Also Read: The Ultimate Guide to What is CAPA Management in Clinical Trials
What Inspection Readiness Requires Across a CRDC Network?
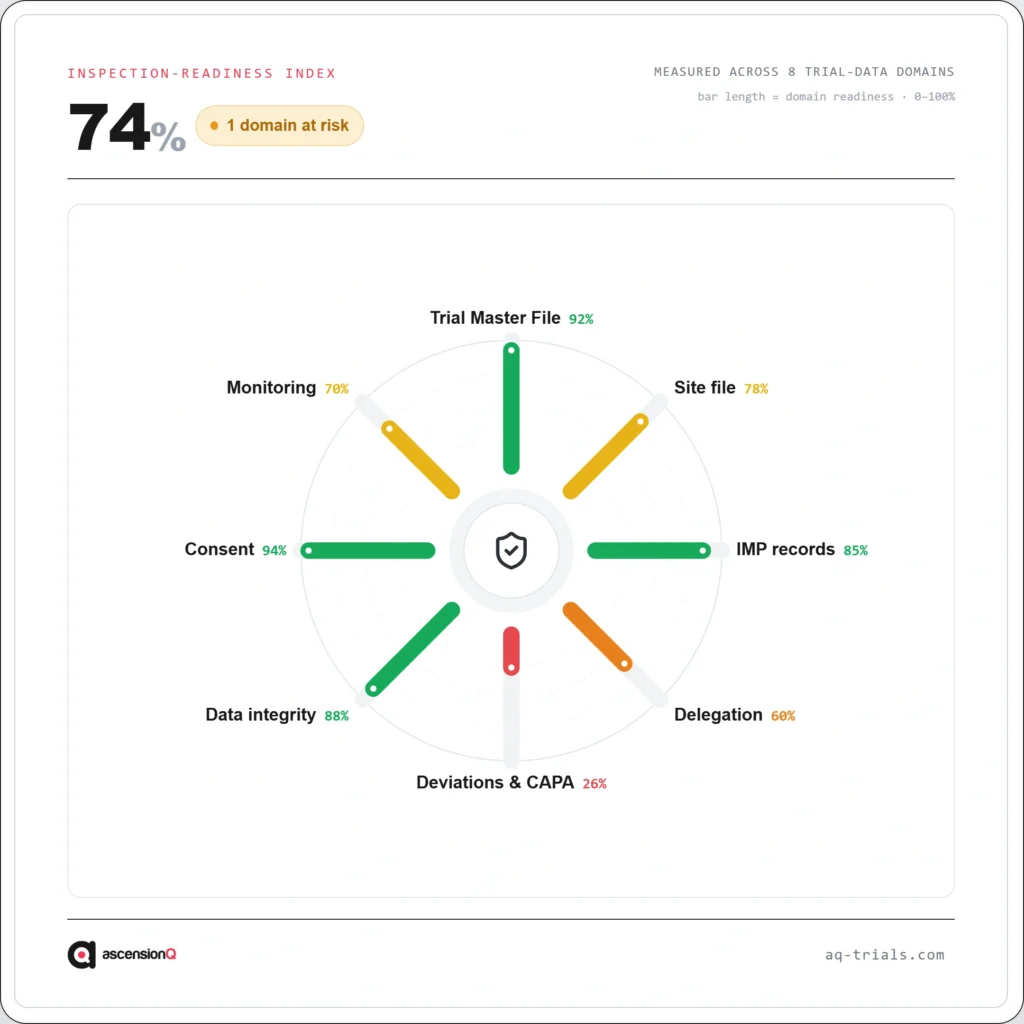
Inspection readiness in a CRDC network is built across five interconnected governance areas. Each must be under active, continuous control. A gap in any one of them creates inspection risk at the hub or at any participating spoke site.
Document Completeness and Version Currency
Every participating site must maintain a complete and current investigator site file. Every document placeholder must be fulfilled by the due date or formally waived with a recorded reason. Every document must reflect the current approved version. Every amendment must be distributed, acknowledged, and filed at each site before the amended procedures take effect.
The hub’s governance responsibility extends beyond issuing documents. It includes confirming that each site received the document, acknowledged it within the required timeframe, and filed it correctly — and maintaining evidence of that confirmation in a form that can be presented at inspection without manual reconstruction.
Delegation of Authority Currency
Current and complete delegation records must be maintained at every participating site throughout the study. Every staff member performing a study task must be listed on the delegation log with their assigned tasks, effective dates, and the Principal Investigator’s authorisation. Updates must be made when roles change, not at the next monitoring visit.
The hub cannot confirm delegation currency at each spoke without access to current delegation records. A hub governance team that relies on monitoring visits to check delegation logs is not exercising continuous governance oversight. It is conducting periodic sampling — which leaves the entire period between visits as an unverified governance gap.
Quality Event Detection and CAPA Resolution
Every deviation, near-miss, and quality event at any participating site must be formally documented, investigated with genuine root cause analysis, actioned with proportionate corrective and preventive measures, and closed with verifiable effectiveness evidence. Hub quality teams must have visibility of quality events across the network as they occur — not when they are escalated by site teams or identified during monitoring.
Patterns across sites must be detectable. A CAPA for an out-of-version consent form at one site and a protocol deviation linked to a recent amendment at another site may share the same root cause: an inadequate amendment distribution and acknowledgement process. The hub quality team can only identify that connection if both events are visible within the same quality governance environment.
Also Read: What is QMS — Quality Management System in Clinical Research?
Pharmacy Documentation and IP Accountability
IP accountability records must be complete, current, and cross-referenceable with the investigator site file at each participating site. Drug receipt, storage, dispensing, and return events must be documented at the time they occur. Temperature excursions must be captured, escalated, and investigated through formal CAPA workflows. Pharmacy authorisations must be current and confirmed.
In a CRDC network, pharmacy documentation at each spoke must be visible to hub governance teams alongside the investigator site file records — not in separate local files that require manual collection before a monitoring visit.
Training and Competency Confirmation
Every staff member who performed a study task must have been trained on the current approved procedure at the time they performed it. Training records must confirm which version of each SOP or procedure was in effect when the training occurred. Acknowledgements of protocol amendments and SOP updates must be documented with timestamps that confirm the staff member had current knowledge before conducting further study activity.
Across a distributed CRDC team, training compliance cannot be confirmed from a hub without structured access to training records at each site. Individual coordinators maintaining their own training logs on spreadsheets cannot provide the hub with the auditable, timestamped evidence that an MHRA inspection requires.
How Fragmented Systems Create Inspection Risk Across a Multi-Site Network?
The governance gaps that produce inspection findings in CRDC networks are not caused by negligent teams. They are caused by structural conditions: documentation managed in disconnected systems, quality events tracked in separate spreadsheets, delegation records maintained in local files, and pharmacy accountability stored in folders that no hub governance team can access without requesting them.
When these structural conditions exist, inspection risk accumulates between monitoring visits whether or not any individual team member is doing their job well. The hub governance team is accountable for oversight it structurally cannot exercise. The spoke site teams are maintaining records that the hub structurally cannot see. The inspection evidence exists in fragments across multiple systems, sites, and file formats — and reconstructing it before an inspection takes days of coordinated effort that the hub may not have when an MHRA notification arrives with five working days’ notice.
Common structural gaps that produce inspection findings in fragmented CRDC networks:
- Protocol amendments distributed by email with no structured acknowledgement tracking — the hub cannot confirm which version each site is operating on without contacting each coordinator individually
- Delegation logs maintained as local files at spoke sites — the hub discovers a gap at the time of a monitoring visit, not at the time the gap appeared
- CAPA records held in site-specific spreadsheets — the hub quality team cannot cross-reference events across sites or identify recurring patterns without manually collating reports from each site team
- Pharmacy documentation in separate local folders — sponsor monitors must request document packages from individual site pharmacists before each monitoring visit, and the hub cannot confirm pharmacy readiness without that process
- Training records updated by site staff on honour systems — completeness cannot be confirmed at hub level without manual chasing, and version currency cannot be verified without cross-referencing the training record against the SOP amendment history
- Overdue document placeholders visible only to the site team — the hub finds out about document gaps when a monitoring visit surfaces them, not when the placeholder first falls overdue
Also Read: What is a Clinical Research Delivery Centre (CRDC) and How Does Multi-Site Research Oversight Work?
What Continuous Inspection Readiness Looks Like in a Connected CRDC?
A CRDC network operating under continuous inspection readiness looks structurally different from one that prepares for each monitoring visit. The difference is not in the diligence of the teams. It is in the operating infrastructure through which governance is exercised.
Document Governance Under Continuous Control
Every participating site operates within a hub-configured electronic investigator site file structure. Document placeholders exist for every required document. Due dates are calculated automatically from milestone reference dates. Overdue alerts reach the relevant site coordinator without the hub having to request a status update. Amendment distribution triggers acknowledgement requirements at every site simultaneously. The hub sees acknowledgement status across all sites in real time — updated as coordinators complete their tasks.
When a document placeholder becomes overdue at any site, the hub governance lead sees it in the same dashboard used for daily oversight. When an amendment is distributed, the hub can confirm within hours which sites have acknowledged it and which have outstanding requirements. The evidence of controlled document governance exists because the governance was exercised continuously — not assembled retrospectively.
Also Read: Electronic Investigator Site File (eISF) in Clinical Research
Delegation Records Confirmed Without Monitoring Visits
Current delegation records at every participating site are accessible to hub governance teams within the same connected environment as the investigator site file. When a role changes at a spoke site, the coordinator updates the delegation record within the platform. The hub governance lead sees the update reflected in the network-wide delegation view without being notified by email or waiting for a monitoring visit.
At any point during the study — including the day before an unannounced inspection — the hub can confirm that every participating site’s delegation records are current, complete, and authorised. The evidence is contemporaneous because it was created at the time of each role change, not assembled in advance of a review.
Quality Events Detected and Actioned at Network Level
Deviations raised at any spoke site are visible to the hub quality team within the connected quality governance environment. CAPAs triggered from document issues, pharmacy events, or deviation records are assigned, investigated, and tracked within a structured eight-stage workflow. The hub quality lead can cross-reference CAPA records across all sites, identify recurring patterns, and extend preventive actions network-wide through the same workflow used to manage the original event.
When an MHRA inspector examines CAPA records at a spoke site, they find structured investigation evidence, documented root cause analysis, specific corrective and preventive actions with named owners and due dates, effectiveness verification by an independent reviewer, and formal closure with full audit trail evidence. The records exist because the quality governance process was followed at the time of each event — not because a team prepared them for the visit.
Also Read: How to Build a CAPA Action Plan That Actually Addresses Root Cause?
Pharmacy Accountability Visible Alongside ISF Records
IP accountability records at each spoke site exist within the same connected environment as the investigator site file. Drug receipt, storage, dispensing, and return records are filed at the time of each event. Temperature excursion records are captured as structured pharmacy events and trigger CAPA workflows directly from the pharmacy record. Storage condition documentation is uploaded and timestamped at the time of each review.
A sponsor monitor preparing for a visit does not request document packages from site pharmacists. The pharmacy records are accessible within the same platform used to review the investigator site file — with full dispensing history, reconciliation records, and IP accountability available alongside document completeness and CAPA status in one governance view.
Also Read: What is ePSF in Clinical Trial Data Management?
Readiness Scores That Reflect Actual Compliance
A cross-site readiness dashboard gives the hub governance lead current visibility of documentation completeness, overdue milestones, signature status, CAPA activity, delegation currency, and pharmacy readiness at every participating site — updated continuously as site teams complete their work.
When an inspection notification arrives, the hub governance lead opens the dashboard, reviews the site’s current compliance status in real time, identifies any outstanding gaps, and coordinates resolution within the available window. The preparation is proportionate because the records were maintained continuously. The visit does not require a filing sprint because the evidence was never allowed to fall into disrepair.
What Each Role Needs to Maintain Inspection Readiness Across a CRDC?
Inspection readiness in a multi-site network is not a governance team responsibility alone. Every role in the network contributes to it. The difference between a network that is continuously ready and one that prepares for each visit lies in whether each role has access to the right information, at the right time, without navigating multiple disconnected systems.
| Role | What Inspection Readiness Requires From This Role |
|---|---|
| CRDC Hub Governance Lead | Real-time visibility of documentation completeness, overdue milestones, CAPA status, and delegation currency across all participating sites — without requiring manual outreach to site teams |
| Research Coordinator (Spoke Site) | Current task list showing required document uploads, overdue placeholders, pending acknowledgements, and signature requirements — without maintaining a parallel tracking spreadsheet |
| Quality Lead (Hub or Site) | Visibility of all open deviations, CAPA stage, root cause investigation progress, and effectiveness verification outcomes across all sites — with the ability to raise CAPAs directly from document records |
| Principal Investigator | Current and authorised delegation records reflecting actual role assignments and effective dates — accessible for review and signature without paper-based log management |
| Pharmacy Staff | Structured IP accountability records within the same environment as the ISF — with dispensing, storage, and return events documented at the time they occur, not collected retrospectively for visits |
| Sponsor Monitor / CRA | Remote access to current study documentation, pharmacy records, CAPA status, and audit trails across sites — without requesting document packages from individual site teams before each visit |
| Research Director / CRDC Lead | Network-wide inspection readiness score, site comparison view, and overdue feed — providing a governance overview that supports accountability without requiring manual status reports from each site |
The Regulatory Foundation of Inspection Readiness in CRDC Research
Inspection readiness in a CRDC network operates within a specific regulatory framework. Understanding each component clarifies why continuous governance control is the only approach that satisfies the standard an inspector applies.
ICH-GCP E6(R3) — Finalised January 2025 and in force in the UK from 28 April 2026, E6(R3) formalises the expectation of quality management systems that enable risk-based, proportionate oversight. It reinforces that essential documents must demonstrate compliance at any point during the trial, not only at scheduled review stages. Continuous inspection readiness is not a higher standard than E6(R3) requires. It is the standard E6(R3) describes.
UK Clinical Trials Regulations (Amendment 2025, in force April 2026) — The most significant update to UK clinical trials regulations in two decades. The new framework reinforces sponsor accountability for oversight across all participating sites, documentation requirements throughout the trial lifecycle, and the expectation that inspection evidence reflects contemporaneous governance activity.
MHRA GCP Inspection Framework — MHRA applies a risk-based approach to inspection selection. Organisations with prior critical or major findings, recent governance changes, or complex multi-site structures face heightened inspection probability. CRDCs — as newly established, multi-site, commercially active research networks — sit within that risk profile. The inspection can come at any time, at the hub or at any spoke site. Readiness built through pre-visit preparation cannot account for that uncertainty.
ALCOA+ — The data integrity standard that governs every record in a clinical trial. Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available. Contemporaneous is the principle most frequently violated by pre-inspection preparation. Records that were reconstructed or compiled in advance of a visit are not contemporaneous. Records created at the time of each activity, filed immediately, and maintained continuously are.
MHRA 25-Year Archiving Requirement — Clinical trial records must be retained for a minimum of 25 years. No document and no quality record can be permanently deleted. Removed items must be archived with full history and restorable by an authorised user. An audit trail that captures this retention evidence from the moment each record is created provides an inspector with a complete, immutable account of the trial’s governance history.
Also Read: What is QMS — Quality Management System in Clinical Research?
How AQ Supports Continuous Inspection Readiness Across a CRDC Network?
AQ provides a connected research operating environment where eISF, ePSF, CAPA, CTMS, Digital DoA, and QMS operate within one platform across every participating site. Hub governance teams maintain real-time visibility of documentation completeness, quality activity, delegation currency, and pharmacy accountability across the network. Spoke site teams work within role-based views that surface their active tasks, overdue requirements, and pending approvals without navigating multiple disconnected systems.

AQ eISF gives the hub a cross-site readiness dashboard showing document completeness, overdue milestones, signature status, and amendment acknowledgement progress at every participating site in real time. Milestone-driven document requirements enforce due dates automatically from site initiation dates. Version control and acknowledgement tracking confirm which document version each site has acknowledged and filed — without relying on email confirmations. Every signature carries a SHA-256 document hash and timestamp, providing tamper-proof evidence for every document across the network.
AQ ePSF brings pharmacy documentation within the same inspection-ready environment as the investigator site file. IP accountability records, dispensing logs, storage condition documentation, and pharmacy authorisations are visible to hub governance teams alongside ISF records in one dashboard view. Temperature excursion records trigger CAPA workflows directly from the pharmacy record, creating a structured quality investigation at the moment of detection rather than after a manual escalation process.
AQ CAPA provides an eight-stage quality investigation lifecycle with formal entry and exit conditions enforced by the system. A CAPA cannot close until an independent verifier records an effective outcome. Every stage transition requires re-authentication and creates an immutable audit trail entry. Hub quality teams can cross-reference CAPA records across all participating sites, identify recurring patterns, and extend preventive actions network-wide within the same workflow used to manage individual site events.
AQ Digital DoA maintains current delegation records at every participating site within a controlled environment visible to hub governance teams. Role assignments, effective dates, and authorisation evidence are accessible in real time without requiring monitoring visits to confirm delegation currency. Delegation gaps surface in the hub dashboard alongside document and quality status — not after an inspector identifies them at site level.
AQ QMS connects controlled documents, SOP governance, training requirements, and staff acknowledgements within one quality layer aligned with daily research operations. Training completion can be confirmed across distributed teams with timestamps linked to the procedure version in effect at the time. SOP acknowledgements are documented as structured records, not tracked by site staff on honour systems.
With AQ, inspection readiness becomes part of your daily research operations, not a last-minute preparation exercise. Documentation, delegation records, CAPAs, pharmacy records, and quality evidence remain current and accessible throughout the study lifecycle. When an MHRA inspection is announced, your team can review cross-site readiness, address any outstanding actions, and demonstrate compliance with confidence.
Visit AQ Features and Comparisons to explore how AQ supports continuous inspection readiness across CRDC networks through connected eISF, ePSF, CTMS, Digital DoA, QMS, and CAPA management. AQ supports modular deployment, allowing your organisation to start with the capabilities you need today and expand into a fully connected research platform over time.
Also Read: What is AQ Platform: Clinical Research Software Modules, Use Cases, and Compliance Standards
Explore AQ Platform for CRDC Inspection Readiness
AQ Trials is an independently accessible module platform within a connected end-to-end clinical research environment, designed to give CRDCs and multi-site research networks the operational control, documentation governance, and quality oversight they need across every participating site.
AQ brings together eISF with multi-site binder management and cross-site readiness dashboard, ePSF for pharmacy documentation and IP accountability within the same inspection-ready environment, CAPA management with structured eight-stage lifecycle and full audit trail evidence, CTMS for end-to-end operational visibility across study execution and site-level milestones, Digital DoA for controlled delegation governance at each participating site, and QMS for connected quality governance with controlled documents, training management, and staff compliance tracking.
Notably, AQ is built around over 20 years of clinical research operations experience and is aligned with ICH-GCP E6(R3), UK Clinical Trials Regulations, MHRA inspection expectations, ALCOA+, 21 CFR Part 11, UK GDPR, Data Protection Act 2018, NHS DSPT, GxP computerised system validation, and GAMP 5.
Book a Live AQ Platform Demonstration or reach out to us for online consultation. Speak with our team to explore how AQ supports CRDC and multi-site research operations through a live walkthrough of hub-and-spoke inspection readiness, eISF, ePSF, and CAPA management across the network.
